Genetics 5 – Cancer Genetics – Flashcards

Unlock all answers in this set
Unlock answersquestion
3 Types of Cancer Genes
answer
1.) Oncogenes 2.) Tumor Suppressor Genes 3.) DNA Repair/Cell Cycle Genes
question
Cancer
answer
Cancer is a disease of the genome. There are 3 different types of genes that are important in the development of cancer: oncogenes, tumor suppressor genes and DNA repair/cell cycle genes. - All "cancer" genes have a function in the normal cell. It is only after activation or inactivation by mutation that the gene promotes development of cancer.
question
Cancer: Inherited vs Somatic Mutations
answer
Both can be important *Somatic mutations*: ast majority of genetic mutations that cause cancer occur in somatic cells and are not inherited *Inherited mutations*: produce families with a high incidence of specific cancers. - However, many of the genes in which mutations cause familial cancer syndromes are also mutated somatically in tumors from non-familial cases!
question
Retrovirus Lifecycle
answer
1.) Retroviruses contain a single RNA strand, copied into DNA by virus-encoded reverse transcriptase (RT) 2.) DNA then integrates into host genome, where it is transcribed by cellular RNA polymerases 3.) Some of the RNA is translated into host proteins, of which there are 3 major genes encoding polypeptides: - *pol - the RT* (which also has integrase activity) - *gag* - forming the *retroviral core* - *env* - the *envelope protein* responsible for host tropism ("Polly Gags with Envy of your Retro style") 4.) The proteins and RNA then assemble into virus particles Sometimes an adjacent gene from host genome is transduced into viral genome; RT tends to mutate these host genes when it gets picked up
question
Weinberg Transformation Assay
answer
Found that the same genes that could be oncogenic in animal retroviruses had also undergone mutation in human tumors. Cancer cells cultured in vitro demonstrate a "transformed" phenotype, exhibiting malignant properties e.g. lack of growth inhibition. Normal cells can also undergo cellular transformation in vitro; clones of cells behaving as such will demonstrate foci in which they pile up on one another - Weinberg extracted DNA from human tumor and "transfected" it into mouse fibroblasts. He found that human gene from the tumor responsible for the focus-forming activity in mouse cells was a mutated version of a gene, Ras, first identified in a retrovirus causing sarcoma in rats Established that it was the oncogenes, rather than any cellular response to the virus, that was leading to cancer
question
Proto-Oncogene
answer
Genes whose normal function is to promote cell growth and proliferation -Proto-oncogenes become oncogenes when mutated or amplified, allowing for unregulated cell growth and differentiation ("Oncogenes call for an On-core/encore")
question
Oncogene
answer
Oncogenes are *dominant*: mutation of only one copy is required to begin the process toward tumor progression. Oncogenes may be activated by any one of several mutational mechanism: - Gene amplification - Activating point mutations - Chromosome rearrangements resulting in upregulation of gene expression - Hypomethylation of the promoter, again allowing increased gene expression *Germline mutations in proto-oncogenes are rare*. Mutations in RET can cause MEN2.
question
Some Oncogenes
answer
A number of oncogenes are now known. Their viral and mutated cellular versions are strongly associated with cancer. (For the viral versions, it is animal cancer, not cancer in humans.) - Src - ErbB - ErbA - Ras - Sis - Fos - Jun - Myc
question
Activation of Proto-Oncogenes by Mutations
answer
*Point Mutation* - RAS missense mutation - Codon 12 or 61 *Translocations* - 9:22 CML Ph chromosomes: Bcr-Abl fusion Protein - 8:14 Burkitt Lymphoma; C-MYC translocated to IgH promoter *Gene Amplification*: - Breast Cancer HER2
question")
Gain of Function Mutations in RET Proto-Oncogene Cause Multiple Endocrine Neoplasia 2 (MEN2)
answer
Proto-oncogenes rarely undergo germline mutation and consequently do not cause heritable cancer syndromes - there are some rare exceptions *RET proto-oncogene*: Germline, heritable RET mutations cause multiple endocrine neoplasia type 2 (MEN2), which predisposes to medullary thyroid carcinoma, parathyroid tumors, and pheochromocytoma
question
MEN1
answer
MEN1, by contrast, is a hereditary syndrome predisposing to a different spectrum of tumors (pituitary, parathyroid, and pancreatic islet) and is caused by mutations in a different gene (a tumor suppressor gene, MENIN).
question
Summary of Oncogenes (lecture summary)
answer
- Identified as transforming genes of retroviruses. - An *activated* form of a cellular gene (*proto-oncogene*). - *Dominant* at cellular level, means only one allele need be mutated. - Mutations are *somatic and never inherited* (an exception being RET). - Retroviruses cause cancer in animals, but not a significant cause of cancer in humans. - Distinct from DNA tumor viruses (i.e. Human Papilloma Virus), in that the transforming oncogene lacks a cellular ortholog, whereas DNA tumor viruses usually have evolved an oncogene entirely of their own (usually called a "T-antigen").
question
Tumor Suppressor Genes
answer
Normal function of tumor suppressor genes is usually to regulate cell division and cell growth, thereby preventing tumor formation - Mutations in tumor suppressor genes are *inherited in an autosomal dominant fashion*. However, the *development of a tumor requires a 2nd (recessive) hit in the cell that goes on to become a tumor* - Mutations in tumor suppressor genes are *loss of function mutations*, as opposed to mutation in *proto-oncogenes, which are gain-of-function mutations*.
question
Tumor Suppressor Genes: Examples
answer
Rb, BRCA1, BRCA2, p53, NF1, APC Germline mutations cause susceptibility to specific cancers depending on which gene is mutated
question")
Loss of Heterozygosity (LOH)
answer
Refers to "loss" of the normal allele once the first one is mutated - originally coined because of the observation that one allele was truly lost (by deletion, for example), leaving only the mutant copy. - Today, we know that the 2nd hit does not have to be deletion, but can be a loss-of-function mutation in the 2nd copy, hypermethylation, etc.
question
Retinoblastoma (Rb) - Review Sheet
answer
Prototypic tumor suppressor gene; led to the "Two-Hit Hypothesis" - early-onset RB tends to be bilateral and have a family history (~40% of cases) - remaining 60% of cases were later onset, unilateral and w/out a family history In *familial cases, the first hit is already present in every single cell* - therefore only one acquired hit (in ANY cell) is required for development of disease. If there is no inherited "first hit", then a given cell must acquire the first hit and then the second hit.
question
Two-Hit Hypothesis
answer
Requirement that both copies of a tumor suppressor gene must be inactivated in order for disease to occur - In familial cases, the first hit is already present in every single cell - therefore only one acquired hit (in ANY cell) is required for development of disease. - If there is no inherited "first hit", then a given cell must acquire the first hit and then the second hit.
question
Retinoblastoma - Lecture
answer
A predominately *pediatric* form of cancer - Retinoblastoma arises from fetal retinoblasts that normally differentiate into post-mitotic retinal photoreceptor cells and neurons and can actually be congenital in onset (i.e. a cancer occurring even at birth)
question
Leukocoria
answer
Seen in *Retinoblastoma* If there's an opacity, such as a tumor or a cataract, blocking the return of light somewhere between the reflective surface of the retina and the cornea, it will show up as the absence of the "red reflex"—leukocoria
question
Retionoblastoma - Associated Tumors
answer
Other tumors associated w/ hereditary retinoblastoma, including: - tumors of the pineal gland of the brain (so-called "trilateral" retinoblastoma) - osteosarcoma of bone In hereditary cases, there can frequently be more than one primary tumor per eye. Orbital irradiation is a form of therapy for the treatment of ocular retinoblastoma. There are case reports where stray radiation induced periorbital osteosarcomas in patients with retinoblastoma.
question
Hereditary vs. Sporadic Retinoblastoma
answer
If *bilateral*, then proportion of *affected offspring ≈ 50% (autosomal dominant* inheritance) ⇒ *HEREDITARY* If *unilateral*, a family history is found in only ≈ 15-20%. So most unilateral tumors are *SPORADIC* 2 Hits for Unilateral Cases & 1 Hit for Bilateral Cases
question
RB Gene and Its Protein
answer
~40% of all cases of retinoblastoma have germline inheritance of RB mutations - DNA-binding *transcriptional repressor* - Interacts with other cell cycle regulators - Regulated by phosphorylation - Somatically mutated in other types of cancer (breast, prostate, CML) that are not associated with retinoblastoma
question
Rb: How come a family history is still found in a minority of unilateral cases?
answer
The probability of a second hit occurring is not certain, because it's still a random event, so there are some patients with germline mutations (as known by family history in parents and children or via genetic testing of non-tumor 'constitutional' tissues, such as blood) who only develop one tumor or sometimes escape cancer altogether. *Most familial forms of cancer caused by mutations in tumor suppressor genes therefore do not have complete penetrance*.
question
Rb: Fairly often the case is bilateral and there is a family history in children of the cases; however the parents are not affected. Why?
answer
*80% of germline cases are new ("de novo") mutations*. This is not surprising. There is *reduced genetic "fitness" because the disease can be lethal*. It is reasonable to expect the incidence of a genetic disease to be constant over time. Consequently, the mutant alleles that are lost because the individuals who have them do not reproduce should be compensated for by new mutations.
question
RB - the *Prototypical "Tumor Suppressor (TS) Gene"*
answer
1.) *TS genes are inherited with autosomal dominant genetics* but *act recessively at the cellular level* 2.) In familial cases, the first hit is inherited and the second hit is somatic. In sporadic cases where the same TS gene is involved, both hits are somatic. 3.) Consequently, cancer is much more likely with hereditary predisposition, and these are its hallmarks: - Earlier age of onset - Multiple primaries (i.e. bilateral involvement). 4.) TS genes are often somatically mutated in many types of cancer, even those that are not encountered in individuals with germline mutations in the gene (i.e. RB mutations in breast cancer, prostate cancer, and CML—which are not associated with hereditary retinoblastoma).
question
p53
answer
Protein is called p53, human gene is TP53 (chrom 17p13) Identified as a target of SV40 T-antigen - SV40 is a DNA tumor virus in animals, probably not harmful to humans; discovered as a contaminant in polio vaccine - SV40 T-antigen "transforms" cells by binding and inactivating p53 "Guardian of the Genome" - *p53 detects DNA damage and either temporarily halts cell cycle so that DNA repair can proceed or initiates apoptotic cell death* ("*P*53 *P*uts a stop to the cell") In addition to its effects on cell growth, *loss of p53 leads to somatic chromosomal abnormalities*
question
Li-Fraumeni Syndrome
answer
Very rare cancer predisposition syndrome in which individuals are at high risk for many types of cancer - Caused by germline mutations in *TP53 or CHK2* ("you might want to sToP and CHecK to see if you have too (frau)Many tumors") - Autosomal dominant inheritance with dependence on somatic 2nd hit. Multiple malignancies: - Breast cancer - Sarcoma - Brain tumors - Leukemia - Adrenocortical carcinoma - Other tumors. Incidence 2×10-5 (rare). Genetic heterogeneity - mutations in *CHK2* cause similar phenotype. CHK2 regulates p53 action.
question")
Neurofibromatosis (NF1)
answer
Autosomal Domonant; Incidence 3×10-4 (not uncommon) - Benign neurofibroma, occasional malignant neurofibrosarcoma, schwanoma, glioma, pheochromocytoma, and certain types of leukemia - "café-au-lait" skin lesions; axillary/inguinal freckling - NF1 encodes neurofibromin (17q11), a GAP (GTPase-activating protein) that down-regulates RAS.
question
Why are the lesions of NF1 punctate? - In other words, how come the skin is not uniformly dark (as opposed to discrete "macules") and why are the neurofibromas scattered in their distribution?
answer
The punctate nature of the lesions (café-au-lait spots and neurofibromas) are visual evidence of the two-hit hypothesis. Loss of function of one allele of NF1 ("haploinsufficiency") appears to have no effect on the cell. It is only when there is bi-allelic loss of both copies of NF1 (the germline loss plus the acquired, somatic second-hit) that cell growth is disturbed in a way that promotes tumorigenesis. Whether the cell without NF1 activity develops into a café-au-lait spot versus a neurofibroma or another type of tumor probably depends upon both the cell of origin that received the second mutation and additional somatic mutations occurring in other genes that also further contribute to tumor growth and oncogenesis.
question")
Familial Adenomatous Polyposis (FAP)
answer
Autosomal dominant disorder caused by mutations in the APC gene - Accounts for ~1% of all cases of colon cancer *APC (5q21)* cytoplasmic protein that interacts with *β-catenin in Wnt signaling* pathway - First hit is germline. Second hit is somatic. - Loss of APC causes *adenoma formation*, then additional mutations (RAS, TP53, SMAD4) transform into full-blown *malignancy* - APC also mutated in sporadic colon adenocarcinoma
question
Familial Adenomatous Polyposis (FAP): Clinical Features and Treatment
answer
Clinical: Polyps begin forming in first decade of life and pts usually have thousands of polyps by late teenage or early adult years Treatment: Prophylactic colectomy in early adult years to avoid inevitable development of adenocarcinoma is the only effective treatment
question
I1307K mutation (KNOW)
answer
present in ~7% of the Ashkenazi Jewish population, increases risk of developing colon cancer but does not cause FAP
question")
Gardner Syndrome (Extra-Colonic Manifestations of FAP)
answer
Even in the era of diagnostic sequencing of the genome, the physical exam remains important. What was at one time called "Gardner Syndrome" represents some of the extra-colonic manifestation of FAP.
question
DNA Repair Genes
answer
2 types of DNA repair genes that are important in the development of cancer: - genes involved directly in *DNA repair* - genes involved in *sensing DNA damage and halting the cell cycle* so that repair can occur
question
Autosomal recessive disorders involving DNA repair gene
answer
- Ataxia Telangiectasia - Fanconi Anemia - Xeroderma Pigmentosum
question
Autosomal Dominant disorders involving DNA repair gene
answer
Heriditary nonpolyposis colorectal cancer (HNPCC)
question
DNA Mismatch Repair Genes
answer
Highly conserved from E. coli through yeast to humans. - Somatic mutations of mismatch repair genes appears important in sporadic cases, because genomic instability of repeated sequences is also noted in many non-familial cases. - A mutation in a DNA repair gene could lead to a *cascade of mutations in many other genes (due to inability to repair mutations)*, including tumor suppressor and proto-oncogenes.
question
Hereditary Nonpolyposis Colon Cancer (HNPCC, aka "Lynch Syndrome")
answer
Predisposition/susceptibility to *numerous cancers, esp of the colon, as well as other GI tract cancers, endometrial cancer and ovarian cancer* - *Autosomal Dominant* - Accounts for ~3-5% of all colon cancer - Tumors exhibit *"microsatellite instability,"* a phenomenon due to *defective mismatch repair* - Genomic instability of repeated sequences had been noted in tumors from pts in HNPCC families. Pattern of instability reminiscent of phenotype in yeast with deficiency in DNA mismatch repair.
question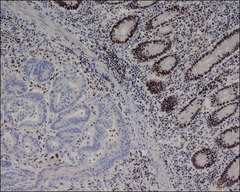
Hereditary Nonpolyposis Colon Cancer (HNPCC, aka "Lynch Syndrome"): Immunohistochemistry (On Review Sheet!)
answer
Immunohistochemistry of the tumor can help direct genetic testing in a pt suspected to have HNPCC - That is, *antibody staining for MLH1, MSH2*, etc. may reveal that *one of the proteins is absent (no staining)* - This is likely because there have been 2 hits - the first germline and the second somatic - and this would be the first gene to test.
question
Hereditary Nonpolyposis Colon Cancer (HNPCC, aka "Lynch Syndrome"): POLYPS (lecture)
answer
A confusing aspect of the name is that it is called "nonpolyposis". Nevertheless, the colon cancers typically do start as benign adenomatous polyps, and individuals with Lynch syndrome probably do have more than their fare share of colon polyps. (And, consequently, endoscopic surveillance screening is hugely important for Lynch syndrome, as removing polyps before they have had a chance to mature into a malignant tumor greatly reduces cancer.) Rather, here, the "nonpolyposis" designation is meant to distinguish Lynch syndrome from FAP, where polyps are found by the boatload.
question")
Somatic Instability in Lynch Syndrome: Microsatellite Instability (Lecture)
answer
Analysis of repetitive DNA sequences (so-called "microsatellite" sequences, containing di-, tri-, or tetra- repeats) upon gel electrophoresis. - The markers themselves showed that they had undergone *heterogeneous mutations leading to gain and loss of a small integral number of repeats * - These patterns were reminiscent of what was known from certain mutants of E. coli that were defective in DNA "mismatch" repair, and the investigators working on this disorder consequently examined the human orthologs of those genes as candidates - Evaluating for microsatellite instability in tumor samples still retains diagnostic utility, as it *can indicate that the individual may have Lynch syndrome and that screening for tumors at other locations may be necessary*
question")
Mismatch Repair Genes in HNPCC (Lecture)
answer
- MSH2 - MSH6 - MLH1 - PMS2 (MySHitty PMS Makes me Lynch Him)
question Pathway")
Schematic of DNA Damage Recognized by the Mismatch Repair (MMR) Pathway
answer
The protein products of the different genes responsible for Lynch syndrome work together in a complex that recognizes and repairs DNA base mismatches.
question
Different types of cancer can occur in different people from the same family with Lynch Syndrome - Why?
answer
First of all, even though we are talking about Lynch syndrome as an example of a DNA repair deficiency syndrome (the third type of cancer gene in our categorization scheme) it can also be thought of as a tumor suppressor gene syndrome. That is, *both copies of the DNA mismatch repair gene* (whether it be MLH1, MSH2, or one of the other genes that can cause Lynch syndrome) *must become mutated* (the first hit inherited through the germline and the second hit occurring somatically). These genes are expressed in many tissues and contribute to DNA repair in many tissues. In and of themselves, the *mutations in the mismatch repair genes are not oncogenic, but rather they lead to subsequent somatic mutations in other tumor suppressor or proto-oncogenes that have a direct role in regulating cell growth.*
question")
Fanconi Anemia (not on disease list!)
answer
A genetically heterogeneous disorder, w/ at least 13 known genes including BRCA2. The disorder is characterized by short stature, radial ray defects, developmental delay and pigmentation defects. Affected pts also have increased risk of leukemia and solid tumors. - Autosomal Recessive (or rarely sex-linked recessive disorder) - Short stature, radial ray limb defects, abnormal pigmentation, developmental delay. - Bone marrow failure. - Increased risk of leukemia and solid tumors (developing at an average age of 23yo).
question
Chromosomal Breakage in Fanconi Anemia
answer
*Breakage Study*: Diagnosis rests on the formation of chromosomal aberrations in cultured cells after treatment with a DNA interstrand crosslinking agent.
question")
Characteristics of the 13 Genes in Fanconi's Anemia (Lecture)
answer
Extreme locus heterogeneity - 12 of the genes are autosomal 1 is on the *X-chromosome*: it is a recessive disorder and is inherited w/ either, most commonly, autosomal recessive or, much less often, sex-linked recessive transmission, respectively - In general, there are no "compound heterozygotes", in that both alleles must be from the same gene - It does not follow a tumor suppressor 2-hit paradigm. (recessive disorder and both alleles mutated in germline (when autosomal recessive)!) Nevertheless, there are still multiple secondary hits in other genes that become mutated consequent to loss of DNA repair activity from the Fanconi DNA repair complex.
question
Fanconi's Anemia and BRCA2
answer
"One of the genes responsible for Fanconia anemia, when you have it in the homozygous state if you are a heterozygous carrier for it you have inherited breast and ovarian cancer syndrom (BRCA2/FANCD1)"
question
Ubquitination
answer
Post-translational modification of protein in which a small polypeptide is ligated to proteins, typically in a oligomeric form, and typically marks them for degradation in the proteasome.
question
The DNA-Repair Pathway in Fanconi's Anemia
answer
All the different FA gene products participate in a common pathway involved in the *recognition and repair of DNA damage*. - An important event in this pathway is the mono-ubiquitination of BRCA2 (FANCD2). - A new clinical test for diagnosis of FA, which complements karyotypes assessing for chromosomal damage in response to DNA interstrand cross-linking agents, is based on *absent mono-ubiquitination of BRCA2*.
question
Hereditary Breast-Ovarian Cancer Syndrome
answer
AD inheritance of either *breast and/or ovarian* cancer in the *same or different individuals* in a family. - Breast/ovarian cancer tend to *occur earlier* and often *recur* or are *bilateral* (as with other TS gene syndromes). - Also associated with *male breast cancer, prostate cancer, and pancreatic cancer*. - Mixture of ancestral and "private" alleles ~2% of *Ashkenazi Jews* heterozygous for 3 ancestral alleles (2 for BRCA1 and 1 for BRCA2).
question
Hereditary Breast-Ovarian Cancer Syndrome: Genes
answer
2 Genes: BRCA1 and BRCA2, both involved in *DNA repair*. - For the most part, follows 2-hit paradigm and BRCA1 and BRCA2 behave as TS genes. - Lifetime penetrance for breast cancer is ~80%, for ovarian cancer ~25-50%.
question
BRCA2: Homozygous vs Heterozygous
answer
*Heterozygous BRCA2 mutations*: causes *autosomal dominant hereditary breast-ovarian cancer syndrome* (which is also caused by mutations in BRCA1). *Homozygous BRCA2 mutations*: cause *Fanconi anemia*! The same mutant BRCA2 alleles can cause either hereditary breast-ovarian cancer syndrome or Fanconi anemia depending upon whether they are present or heterozygously, respectively.
question
Next-Generation DNA Sequencing Gene Panels
answer
Many genes are associated w/a variety of inherited cancer predisposition syndromes; some primarily associated w/ a specific type of cancer, others more generally predisposed to cancer in tissues throughout the body. Some disorders have additional features, not obviously connected to cancer predisposition (e.g., macrocephaly in Cowden syndrome due to PTEN mutations). - Increasingly, genetic testing employs *large, multigene panels that use 'next-generation' DNA sequencing technologies to rapidly screen large numbers of genes*. One such panel known as the 'BROCA' panel
question
Common Variants
answer
Several distinguishing features: - common in the general population - as a rule, tend to not be as harmful - 'penetrance' is low - In contrast, genuine 'Mendelian' mutations tend to be highly penetrant. Common variants are often outside of a coding region of a gene, where they may influence gene expression.
question
Common Variants and Cancer Risk
answer
Common Variants Also Contribute to Cancer Predisposition: - Some 'common' variants are also associated w/ cancer risk, in this case, an intronic regulatory polymorphism in the RAD51B DNA repair gene, for which I happen to be homozygous.
question
What's the difference between cancer predisposition resulting from a so-called common variant and a Mendelian-form of cancer?
answer
Have several distinguishing features. First of all, they are common in the general population, whereas what we think of as single-gene mutations responsible for cancer predisposition tend to be rare and unique to a family (or else somewhat more common in a particular population due to a 'founder effect'). - Common variants, as a rule, tend to not be as harmful, and their 'penetrance' is low. Perhaps if they were, they would be selected against and might not rise to such a high level in the population. And under other circumstances, they may have even been beneficial, contributing to their selection within a population. - In contrast, genuine 'Mendelian' mutations tend to be highly penetrant. Common variants are often outside of a coding region of a gene, where they may influence gene expression. - In contrast, the mutations responsible for highly-penetrant single-gene disorders tend to be within coding sequences and have major effects disrupting the function of an encoded protein. Because common variants have low penetrance, it may not be obvious that they run in a family, whereas a highly penetrant, autosomal-dominant cancer predisposition syndrome is more likely to show the signature multigenerational pattern of inheritance. Because of the difference in penetrance, different approaches to gene discovery are required. For highly penetrant single-gene disorders, it is possible to track inheritance within a family based on phenotype and thereby identify the causative gene via genetic linkage (in the past) or genomic/exomic sequencing (modern era). In contrast, for common variants, a population level approach, employing genetic association (i.e., GWAS (genome-wide association study)) is usually required.
question
Are common variants inherited in a Mendelian fashion?
answer
Yes, of course, the rules of Mendelian inheritance apply to all nuclear genes.
question
A patient comes to you with questions regarding a 23andMe testing result for a common variant; what are some things to keep in mind?
answer
In sum, take it with a grain of salt. First of all, many GWAS variants have not been validated in confirmatory studies, suggesting that they could represent publication bias (though 23andMe does a reasonably good job of indicating how robust the data are). Second, variants identified through association studies are highly population dependent, because the common variant arises on a particular ancestral haplotype (extended chromosomal region from a common ancestor). Consequently, the particular variant is not necessarily causal by itself, but may merely just 'tag' a haplotype containing other variants that by themselves, or collectively, influence disease risk. As a result, *different populations will have different haplotypes* (and different linked variants) associated with this variant. *GWAS results tend only to be applicable to the same population in which the study was performed*. They are particularly *difficult to interpret in people who have multiracial ancestry*. In most cases, the disease risk conferred by a common variant is much less than is the case for a mutation in a highly-penetrant gene responsible for a Mendelian cancer predisposition syndrome. Finally, *common variants tend to act additively*, and it is the sum of these weakly acting risk factors that influence disease predisposition. So a single higher risk variant may be counter-balanced by a combination of lower-risk variants. And, depending upon the strength of their effects, not all of these risks may have yet been discovered.
question
Xeroderma Pigmentosum (not on disease list!)
answer
Also genetically heterogeneous - Characterized by extreme sun sensitivity and high risk of developing skin cancer - The DNA repair defect in XP results in an inability to repair UV-induced thymine dimers by the excision repair pathway.

